Peptide mass fingerprinting, Tandem mass spectrometry (ms/ms), Establishment of 2-d databases – Bio-Rad GS-900™ Calibrated Densitometer User Manual
Page 38
m/z
m/z
MS
MS/MS
Ion current
Ion current
AWGI
SPVR
AWGIS
ISPVR
AWGISP
AWGISPV
VQVSR
AWGISPVR
QGLWIVDMSSGAVK
NQNEYQVSWDTEK
ENIYPEDQQESPSIGLK
WGISPVR
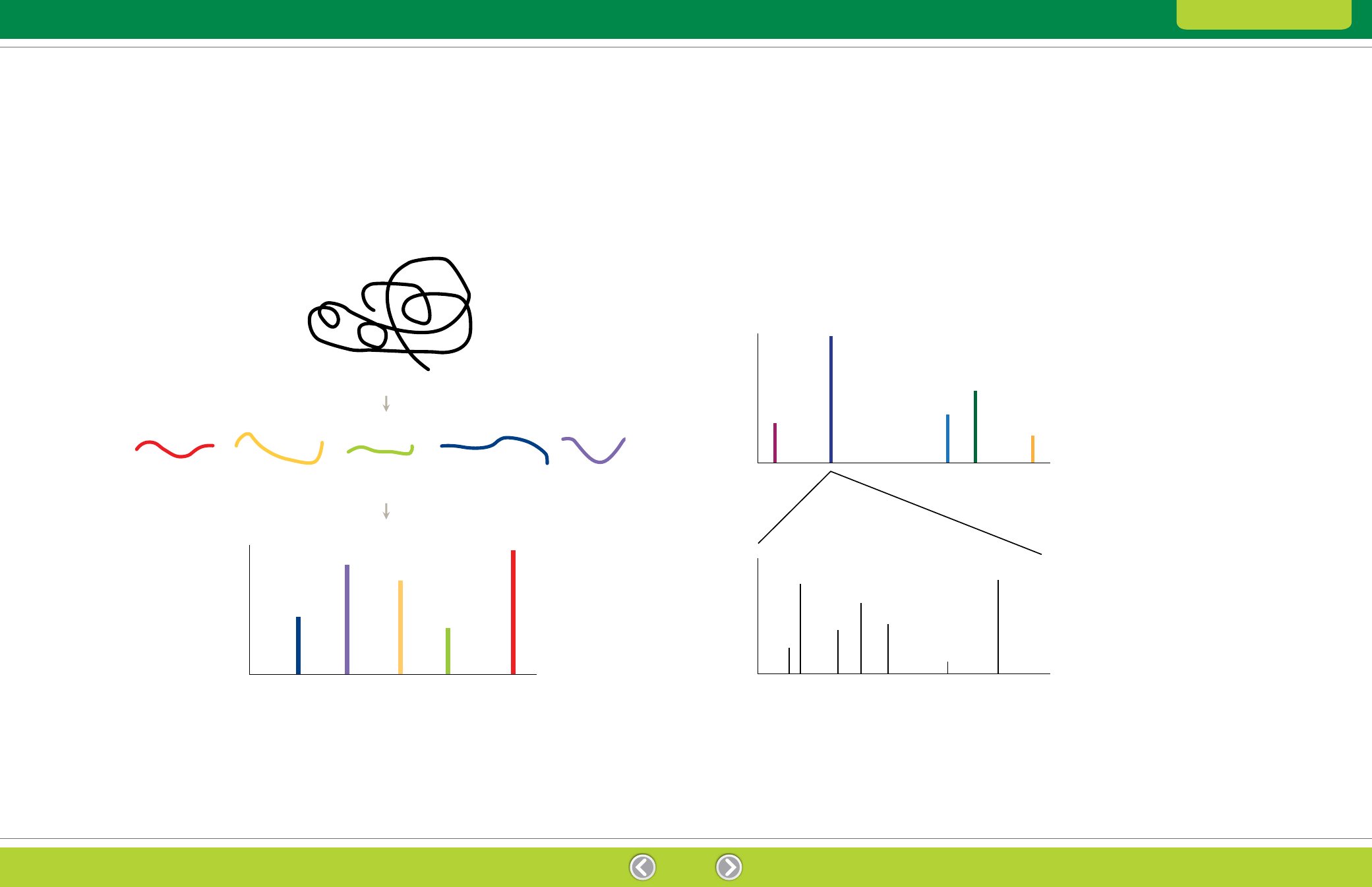
Fig. 7.3. MS/MS analysis. The first mass analyzer selects ions of a particular m/z for fragmentation. The second mass analyzer produces the
mass spectrum for those fragments.
Fig. 7.2. Peptide mass fingerprinting. Peptides resulting from digestion are analyzed by mass spectrometry, and the resulting m/z values
and mass spectrum are compared to theoretical values derived from “in silico” digestion of known proteins in a database.
N
C
N
C
C
C
N
N
N
C
m/z
Abundance
N
C
Analysis
Digestion
72
73
2-D Electrophoresis Guide
Theory and Product Selection
Chapter 7: Identification and Characterization of 2-D Protein Spots
of peptide mass fingerprinting, however, include the
following: (i) the protein sequence has to be present
in the database of interest, and (ii) several peptides
are required to uniquely identify a protein. Additionally,
most algorithms assume that the peptides come from
a single protein, which is why resolution in the 2-D
separation is so critical. If this information does not
allow unequivocal identification of the protein, peptides
can then be analyzed by tandem mass spectrometry.
Peptide Mass Fingerprinting
In this method, the peptides resulting from digestion
of the protein of interest are analyzed by mass
spectrometry and compared to a database of
calculated peptide masses generated by “in silico”
cleavage of protein sequences using the same
specificity as the enzyme that was employed in the
experiment. Identifications (“hits”) are scored in terms
of confidence of match (Figure 7.2).
This approach requires simple mixtures of proteins or
pure proteins and is, therefore, suitable for analysis of
proteins isolated from 2-D electrophoresis. Limitations
Tandem Mass Spectrometry (MS/MS)
In MS/MS, a peptide ion is isolated in the mass
analyzer and subjected to dissociation to product ion
fragments. Peptides dissociate according to certain
rules. For example, fragmentation typically occurs
along the peptide backbone; each residue of the
peptide chain is successively cut off, both in the N->C
(a-, b-, c- ions) and C->N (x-, y-, z- ions) directions.
The product ions resulting from the fragmentation
are analyzed in a second stage of mass analysis,
which enables sequence derivation (Figure 7.3).
Tandem MS can allow identification of proteins
from a single peptide (Lovric 2011).
Establishment of 2-D Databases
After the spots are cut, analyzed, and identified, by
MS for example, the information can be imported
back into the experiment as annotations. Annotations
are organized in categories, for example by protein
name, protein family amino acid composition,
protein function, cellular location, binding properties,
and translational regulation. A single spot may be
annotated in multiple categories, depending on the
amount and type of information available about it.
Most categories contain simple text annotations.
Specialized categories can be used to link spots
to Internet protein databases or to open files in
other applications.