Beyond excision, Proteolytic digestion, Washing – Bio-Rad GS-900™ Calibrated Densitometer User Manual
Page 37: Reduction and alkylation, In-gel proteolytic digestion, Identification by mass spectrometry
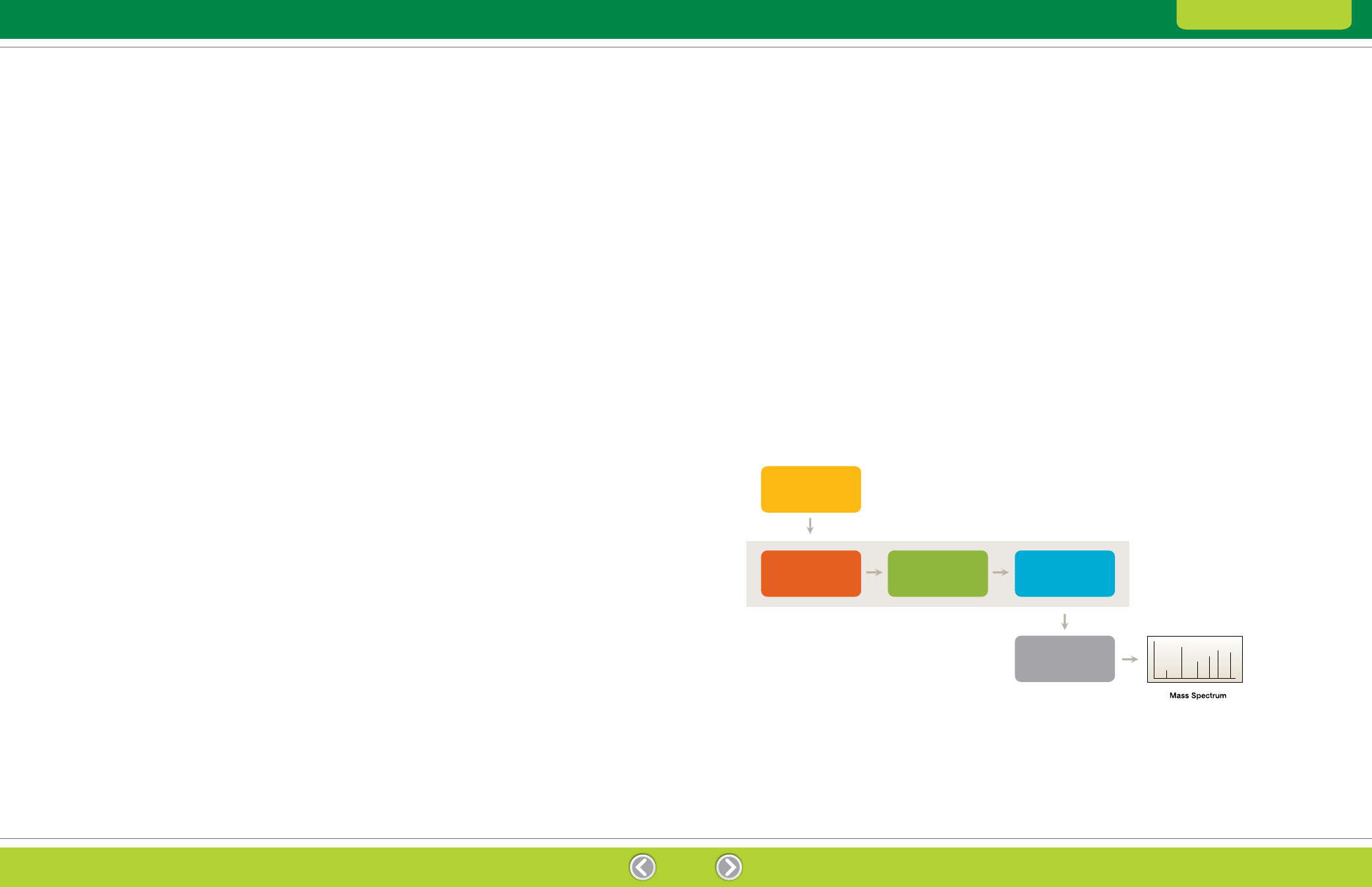
Fig. 7.1. Components of a mass spectrometer.
Sample
Introduction
Ionization
Source
Mass
Analyzer
Detector
Data
Handling
70
71
2-D Electrophoresis Guide
Theory and Product Selection
Chapter 7: Identification and Characterization of 2-D Protein Spots
In-Gel Proteolytic Digestion
Proteolytic digestion can be performed directly on
processed gel pieces. Because proteases are also
subject to autolysis, always include a blank piece as a
control. Proteases used for this purpose are selected
for their efficiency in in-gel digestion and for their
defined cleavage specificity, which allows prediction of
the generated peptide masses. The most commonly
used protease is trypsin, but other proteases used
include LysC, GluC, ArgC, AspN, and LysN, which
cleave to either the C- or N-terminal side of a single
amino acid, as signified by their nomenclature.
These enzymes are all commercially available as
preparations that have been specifically modified for
use prior to mass spectrometry. Enzymes specifically
recommended for mass spectrometry should always
be used for in-gel digestion.
■
■
Use trypsin (modified porcine pancreatic trypsin,
mass spectrometry grade) for initial protein digestion.
Trypsin is one of the most specific proteases and
cleaves at the C-terminal side of Arg and Lys
■
■
Use GluC, AspN, or LysC with proteins of smaller
mass. These enzymes generate fewer peptides
of larger mass than trypsin, which may generate
fragments too small for definitive identification
■
■
Use acid hydrolysis, cyanogen bromide cleavage,
or other chemical methods if alternatives to
enzymatic digestion are required
■
■
Some proteins are processed forms of larger
proteins; therefore, once the protein is identified
based on a trypsin digestion, other methods can be
used to define the N- and C-termini of the fragment
The resulting peptides can be extracted with
acetonitrile, dried under vacuum, and dissolved in a
small amount of water. Prior to mass spectrometry,
the samples should be further purified by solid phase
extraction, for example using ZipTip pipet tips.
A protocol is provided in Part II of this guide.
Beyond Excision
2-D electrophoresis has the unique capability of
simultaneously displaying several hundred proteins.
When coupled with the ability of mass spectrometry
to identify and characterize small quantities of protein,
2-D electrophoresis is a very powerful and effective
analytical method.
Several mass spectrometric techniques can be
used for protein identification at the end of a 2-D
electrophoresis workflow. Most of these methods first
require proteolytic digestion of the protein into discrete
fragments that can be eluted from the excised gel
plug. The most basic mass spectrometric method,
peptide mass fingerprinting, simply determines
accurate masses of the peptides generated.
These masses are then compared to a database,
and the protein of origin can often be uniquely
identified. Another technique, tandem mass
spectrometry (MS/MS) further fragments selected
peptides along the peptide backbone, allowing the
generation of limited sequence information that can
be used to refine the protein identification step.
Proteolytic Digestion
In-gel digestion (Rosenfeld 1992) of selected proteins
is part of the sample preparation process for mass
spectrometry, and it comprises four basic steps:
destaining (washing) the gel pieces, reduction and
alkylation, proteolytic cleavage of the protein,
and extraction of the resultant peptides.
Washing
After excision of the protein spot of interest from the
gel, most protocols require destaining of the proteins
before proceeding. The destaining or wash protocol
depends on the stain used for visualization.
Commonly used protocols for various stains are
described in Part II of this guide.
Reduction and Alkylation
Reduction and alkylation together reduce and
irreversibly block the formation of inter- and
intramolecular disulfide bridges, which can significantly
improve the efficacy of proteolytic cleavage and
subsequent mass spectrometry.
Proteins excised from 2-D gels have usually been
reduced and alkylated either during sample
preparation or equilibration prior to the second
dimension and may not require this step. This step
is mandatory if upstream processing did not
incorporate reduction and alkylation. Any reduction
or reduction plus alkylation step must be followed
by a cleanup step prior to mass spectrometry.
Identification by Mass Spectrometry
Identification of the peptides derived from digestion
can be achieved using several mass spectrometry
techniques. Only a brief overview of mass
spectrometry theory and techniques is presented here.
Refer to the literature from mass spectrometer vendors
for more information about systems and methods.
Mass spectrometry systems contain the following
components (Figure 7.1):
■
■
Ionization source — converts the sample into
gas-phase ions, which are then injected into a mass
analyzer. The two ionization sources most commonly
used for peptide mass spectrometry are matrix-
assisted laser desorption ionization (MALDI) and
electrospray ionization (ESI)
– MALDI — the protein is mixed with an organic
molecule (the “matrix”), deposited onto a planar
substrate, allowed to dry, and illuminated with a
pulsed UV laser. The matrix compound absorbs
the laser energy and promotes peptide ionization,
typically generating singly-charged molecular ions.
MALDI is useful for high-throughput applications
but is limited by ion suppression (particularly in
complex peptide mixtures) and chemical noise
from the matrix in the low mass range
– ESI — a flowing liquid is passed through a
charged orifice to produce charged droplets,
which are then desolvated to yield gas-phase
peptide ions. ESI can be coupled directly to liquid-
phase separations such as chromatography
(LC-MS) and generates multiply-charged
molecular ions that bring mass-to-charge ratio
(m/z) values within the mass range of mass
spectrometry instruments most commonly
used with ESI
■
■
Mass analyzer — sorts the ions according the m/z.
A number of different types of mass analyzers are
available, including time-of-flight (TOF), quadrupole,
and ion trap systems as well as combinations of
these (hydrid mass spectrometers)
■
■
Ion detector — records the ion current, amplifies it,
and sends it to the data analysis system where it is
presented in the form of a mass spectrum. The m/z
values of the ions are plotted against their intensities
to show the number of components in the sample,
the molecular mass of each component, and the
relative abundance of the various components in
the sample
The data from the mass analyzer(s) are used for protein
identification, and two options are most common
in the 2-D electrophoresis workflow: peptide mass
fingerprinting and tandem mass spectrometry.